
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
Психология и педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Рефераты по сексологии
Рефераты по информатике программированию
Краткое содержание произведений
Реферат: Влияние физических и химических факторов на основность алкиламинов
Реферат: Влияние физических и химических факторов на основность алкиламинов
Кислоты и основания
Протон наиболее естественно воспринимается химиком как катион атома водорода"
Р.Белл
В современной химии используются две теории кислот и оснований: теория Бренстеда - Лоури и теория Льюиса - Усановича. Более общие определения кислот и оснований предложены в теории Льюиса - Уеановича. Однако в связи с важной ролью про-толитических процессов в химии теория Бренстеда - Лоури сохранила самостоятельное значение. Именно этой теории мы обязаны появлением комплекса проблем, ставших предметом настоящей книги.
1.1. Теория Бренстеда - Лоури
Согласно теории Бренстеда - Лоури кислота рассматривается как вещество, поставляющее протон, а основание - как вещество, способное присоединять протон (1,2)
![]() А
В +Н + (1)
А
В +Н + (1)
Кислота и отвечающее ей основание, таким образом, образуют сопряженную пару.
Ключевым в теории Бренстеда - Лоури является представление о том, что кислота взаимодействует при переносе протона с другой сопряженной парой (двойное протолитическое равновесие):
![]() А1+
В2 А2 + В1 (2)
А1+
В2 А2 + В1 (2)
Отсюда вытекают и первые предпосылки для количественного сравнения силы кислот и оснований: сильная кислота отдает протон ”легче”, чем слабая, а сильное основание “крепче” связывает протон, чем слабое. Равновесие тем полнее сдвинуто вправо, чем сильнее кислота А1 и слабее кислота А2 .
Можно представить, что двойное протолитическое равновесие является результатом двух сопряжённых равновесий:
![]() НА Н+ + А- (3)
НА Н+ + А- (3)
![]() В + Н+ ВН+
(4)
В + Н+ ВН+
(4)
Уравнение (4) лежит в основе количественной теории .позволяющей сравнивать силу различных оснований.
Безусловно, схема кислотно-основного равновесия, сформулированная Бренстедом и Лоури, не полностью отражает истинный процесс. Вернее, уравнение (4) следует считать адекватным процессам, происходящим в газовой фазе, что само по себе достаточно ценно и важно. В растворах же реакция между кислотой и основанием не сводится только к переносу протона от кислоты к основанию. Сначала кислота АН и основание В образуют комплекс АН...В за счет водородной связи между водородом кислоты и электронодонорным атомом основания. Во многих случаях протолитическая реакция преимущественно ограничивается этой начальной стадией комплексообразования. Поэтому такой процесс называется "незавершенным" кислотно-основным равновесием [4]. Следовательно, образование водородных связей рассматривается не только как вспомогательная, переходная ступень при кислотно-основном взаимодействии, облегчающая переход протона от кислоты к основанию, но и как один из самостоятельных видов этого взаимодействия. В благоприятных условиях кислотно-основное взаимодействие не останавливается на стадии комплекса АН...В, и происходит передача протона от кислоты к основанию, в результате чего основание протонируется. Эта вторая стадия цротолитического процесса называется "завершенным" кислотно-основным взаимодействием. При этом образовавшиеся ионы могут находиться в растворе либо в свободном виде, либо в виде ионных пар:
![]()
![]() ВН+А- ВН+ | | А- ВН+ + А-,
(5)
ВН+А- ВН+ | | А- ВН+ + А-,
(5)
где ВН+А- - тесные ионные пары; ВН+ ççА-- сольватно-разделеные ионные пары ; ВН+ и А--свободные ионы.
Более полным отражением кислотно-основного процесса является следующая схема [4]:
![]()
![]()
![]() АН
+ В а АН...В б А-...ВН+ в А- + ВН+,
(6)
АН
+ В а АН...В б А-...ВН+ в А- + ВН+,
(6)
здесь а - незавершенное кислотно-основное равновесие, б - завершенное и в - диссоциация на свободные ионы*. Таким образом, количественное сравнение слабых оснований в рамках теории Бренстеда - Лоури должно осуществляться с учетом реальной ситуации, в которой происходит перенос протона к органическому основанию: в газовой фазе основой для количественных расчетов основности может служить принципиальная схема (4), тогда как в растворах следует опираться на схему (6), а вернее, на ее модификации, учитывающие конкретные условия.
1.2. Физический смысл и меры основности в газовой фазе
Основностью в газовой фазе называют свободную энергию (DG) равновесия (4) [5,6] . Как известно, DG = DН0 - ТDS. Измерения энтропии равновесия в газовой фазе показали, что эта величина обычно не превышает 9-12 Дж/(моль·К). Таким образом, изменение энтальпии равновесия (DН0) считается равным (DG0) [5].
Изменение энтальпии равновесия (4) переноса протона в газовой фазе, взятое с обратным знаком, называется сродством к протону и обозначается РА (Proton Affinity). Численное значение РА определяется из соотношения:
РА=-DН0 =DН0 (Н+ )+ DН0 (В) - DН0 (ВН+) (7)
Где DН0 (Н+) - энтальпия образования иона Н+; DН0 (В) и DН0 (ВН+) - энтальпия образования основания (В) и его протонированной формы (ВН+) соответственно. DН0 (Н+) = 1530 кДж. Энтальпии образования органических соединений известны из термохимических справочников или могут быть вычислены по соответствующим эмпирическим формулам [7]. Следовательно, определение сродства к протону РА зависит от возможности определения энтальпии образования протонированной формы основания DН0 (ВН+).
Экспериментально определение сродства оснований к протону осуществляется с помощью спектроскопии ионного циклотронного резонанса [8] и масс-спектрометрии высокого давления [5. В настоящее время эти методы позволяют вычислять значения РА с точностью ±0,9 кДж/моль. Таким образом, величина РА является надежным критерием для сравнения основности соединений в газовой фазе.
Существует мнение [5], что на базе значений РА можно построить ”абсолютную”” шкалу основности органических соединений. В последние годы получены значения РА для некоторых алифатических и ароматических аминов, гетероциклических соединений, спиртов, эфиров, альдегидов, кетонов, карбоновых кислот [5] и нитросоединений [9].
2. Основность аминов в газовой фазе
Из рассмотрения данных по основности аминов в растворах следует, что она существенно зависит от электронных и пространственных факторов структуры и сольватации исследуемых соединений. Несмотря на отмеченные успехи при выявлении отдельных зависимостей между основностью и указанными эффектами, до сих нет единого количественного подхода к решению проблемы в целом. Казалось бы, что для оценки влияния строения аминов на их основность наиболее удобными есть соответствующие характеристикистики в газовой фазе, в которой, естественно, сольватационные эффекты отсутствуют. С этой точки зрения большой интерес представляет серия появившихся в последнее время работ (в том числе и обзорных [3, 6, 7, 21,137—140]), в которых обобщены результаты масс-спектрометрических методов, в которых определялись энергии перехода протона между стандартным и рассматриваемым (В) аминами в газовой фазе:
![]() В0Н+
+ В В0 + ВН + (1)
В0Н+
+ В В0 + ВН + (1)
Наличие свободной энергии этого процесса характеризует основность амина В относительно стандарта В0. Абсолютная основность (В0), равная отрицательной свободной энергии (DG0) процесса (8), может быть рассчитана, если известно соответствующее
В + Н+=ВН+ (2)
абсолютное значение для стандартного основания. Энтальпия (DH0) процесса (8), взятая с обратным знаком, характеризует сродство аминов к протону (РА) и легко рассчитывается, так как для прото-нирования аммиака, обычно принимаемого за стандарт, она определена независимыми методами [139, 143, 148, 149, 150]. Абсолютные значения 0В и РА* зависят от выбранного стандарта и абсолютной величины РА для него (например, для аммиака значение РА изменяется достаточно широко: 200,7 [139]; 201,4 [151]; 202,3 ± 2,0 [152]; 207 ± 3 [143, 148, 150, 153], 211,3 [149]; 214,4 [45]). Поэтому для выяснения количественных закономерностей влияния структуры аминов на их основность в газовой фазе лучше всего пользоваться величинами DGB (или — dRDG0 [3,7, 47]), когда за стандарт выбран аммиак**.
В табл.1 приведены величины DGB известного к началу 1977 г. ряда аминосоединений. Сопоставление этих данных показывает, что поведение различных аминов как оснований в газовой фазе резко отличается от такового в конденсированных средах. Например, анилин (табл. 1, № 31) в газовой фазе оказался на 6,7 ккал/моль (или почти на 5 ед. рКа) более основным, чем аммиак (№ 1), в то время как в воде, нитрометане и ацетонитриле (см. выше) наблюдается противоположная ситуация. Аналогичная картина имеет место и для пиридина, который в конденсированной фазе примерно на 4 ед. рКа менее основен, а в газовой фазе (№ 80) на 16 ккал/моль (или ~ на 12 ед. рКа) более основен, чем аммиак; для ацетамида (№ 32), который в воде ~ на 10 ед. рКа менее основен по сравнению с аммиаком, а в газовой фазе практически равен ему; для пиррола (№ 54) и некоторых других аминосоединений.
Таблица 1. Значения основностиа аминосоеденений в газовой фазеб относительно аммиака
| Номер | Амин |
DGBг) |
||||
| 1 |
NH3 |
0,0 | ||||
| 2 |
CH3NH2 |
9,1 | ||||
| 3 |
C2H5NH2 |
11,8 | ||||
| 4 |
n-C3H7NH2 |
13,0 | ||||
| 5 |
i-C3H7NH2 |
14,1 | ||||
| 6 |
n-C4H9NH2 |
13,5 | ||||
| 7 |
i-C4H9NH2 |
14,0 | ||||
| 8 |
s-C4H9NH2 |
15,2 | ||||
| 9 |
t-C4H9NH2 |
16,1 | ||||
| 10 |
n-C5H11NH2 |
13,4д) |
||||
| 11 |
t-C5H11NH2 |
17,4 | ||||
| 12 |
n-C6H13NH2 |
13,5д) |
||||
| 13 |
n-C7H15NH2 |
13,6д) |
||||
| 14 |
c-C6H11NH2 |
16,3 | ||||
| 15 |
NH2-NH2 |
3,8 | ||||
| 16 |
NH2(CH2)2NH2 |
19,0д) |
||||
| 17 |
NH2(CH2)3NH2 |
24,5д) |
||||
| 18 |
NH2(CH2)4NH2 |
27,1д) |
||||
| 19 |
NH2(CH2)5NH2 |
25,4д) |
||||
| 20 |
NH2(CH2)6NH2 |
25,4д) |
||||
| 21 |
NH2(CH2)7NH2 |
25,2е) |
||||
| 22 |
CH3O(CH2)2NH2 |
14,7д) |
||||
| 23 |
H2C=CH-CH2NH2 |
11,3 | ||||
|
|
HCºC-CH2NH2 |
6,7 | ||||
| 25 |
NCCH2CH2NH2 |
3,0 | ||||
| 26 |
CF3(CH2)3NH2 |
10,1 | ||||
| 27 |
FCH2CH2NH2 |
8,0 | ||||
| 28 |
CF3(CH2)2NH2 |
6,7 | ||||
| 29 |
F2CHCH2NH2 |
4,0 | ||||
| 30 |
CF3CH2NH2 |
-1,4 | ||||
| 31 |
|
6,8 | ||||
| 32 |
CH3CONH2 |
0,2ж) |
||||
| 33 |
HCONH2 |
-7,1з) |
||||
| 34 |
(CH3)2NH |
15,5 | ||||
| 35 |
CH3NHC2H5 |
17,9 | ||||
| 36 |
(C2H5)2NH |
20,2 | ||||
| 37 |
(n-C3H7)2NH |
22,2 | ||||
| 38 |
(i-C3H7)2NH |
23,9 | ||||
| 39 |
(n-C4H9)2NH |
23,1 | ||||
| 40 |
(i-C4H9)2NH |
23,6 | ||||
| 41 |
(s-C4H9)2NH |
25,8 | ||||
| 42 |
|
11,2 | ||||
| 43 |
|
18,0 | ||||
| 44 |
|
20,1 | ||||
| 45 |
|
21,2 | ||||
| 46 |
|
19,2д) |
||||
| 47 |
|
14,4д) |
||||
| 48 |
(H2C=CHCH2)2NH |
19,3 | ||||
| 49 |
(HCºCCH2)2NH |
11,7 | ||||
| 50 |
NCCH2NHCH3 |
2,7з) |
||||
| 51 |
CF3CH2NHCH3 |
6,2з) |
||||
| 52 |
|
12,9 | ||||
| 53 |
|
15,3ж) |
||||
| 54 |
|
4,0ж) |
||||
| 55 |
NHMe-C=O H |
1,7 | ||||
| 56 |
(CH3)3N |
20,0 | ||||
| 57 |
(CH3)2NC2H5 |
22,4 | ||||
| 58 |
(C2H5)2NCH3 |
24,6 | ||||
| 59 |
(C2H5)3N |
26,7 | ||||
| 60 |
(C3H7)3N |
28,7 | ||||
| 61 |
|
17,1и) |
||||
| 62 |
|
8,1и) |
||||
| 63 |
|
24,3 | ||||
| 64 |
|
25,7 | ||||
| 65 |
|
27,1 | ||||
| 66 |
|
26,1к) |
||||
| 67 |
(CH3)2N-NH2 |
15,2 | ||||
| 68 |
((CH3)2NCH2)2 |
30,3 | ||||
| 69 |
|
23,5 | ||||
|
|
||||||
70 |
(H2C=CH-CH2)3N |
24,7 | ||||
| 71 |
(HCºC-CH2)3N |
15,0 | ||||
| 72 |
NCCH2N(CH3)2 |
7,1 | ||||
| 73 |
F3CCH2N(CH3)2 |
20,9 | ||||
| 74 |
|
19,5 | ||||
| 75 |
|
21,0ж) |
||||
| 76 |
|
23,7ж) |
||||
| 77 |
|
19,3 | ||||
| 78 |
|
21,8 | ||||
| 79 |
|
26,0 | ||||
| 80 |
|
16,0 | ||||
| 81 |
CH3CON(CH3)2 |
11,7 | ||||
| 82 |
HCON(CH3)2 |
7,6 | ||||
| 83 |
NF3 |
-56л) |
Существенные различия между свойствами в газовой и конденсированной фазах наблюдается и при сравнении оснований одного и того же класса. Например, все первичные алкиламины в газовой фазе (№2—29), за исключением b,b,b-трифторэтиламина (№30), оказались более основными, в то время как в воде (см. например, табл. 1) амины с электроотрицательными заместителями зачастую менее основны, чем аммиак. То самое относится и ко вторичным и третичным алкиламинам.
Данные по изменению свободной энергии и энтальпии реакций, описываемых уравнениями (17) — (2), совместно с некоторыми другими результатами позволили определить термодинамические характеристики процессов переноса свободных и протонированпых оснований из газовой фазы в водные растворы и на этой основе про вести термодинамический анализ влияния сольватации на основность аминов в воде [3, 6, 47, 140, 151, 153]. При этом преимущественное внимание было уделено причинам, обусловливающим наблюдаемый порядок изменения основностии в воде при переходе от аммиака к первичным, вторичным и третичным алкиламинам с насыщенными углеводородными заместителями. На основе этих данных Ариетт с сотрудниками [3, 6, 47] сделал вывод, что главным фактором, определяющим наблюдаемый порядок основности аминов различных классов в воде, является специфическая сольватация соответствующих катионов, зависящая от числа атомов водорода у протонированного азота. Неспецифическая же сольватация, по их мнению, имеет второстепенное значение, т. е. эти исследователи придерживаются сольватационной (гидратационной) теории Тротмана — Диккенсона (см. выше).
В то же время другая группа исследователей [140, 153] считает, что изменение основности аминов при переходе из газовой фазы в воду обусловлено прежде всего электростатической (неспецифической) сольватацией катионов, а специфическое взаимодействие играет второстепенную роль. При этом в указанных работах принимается, что кислотно-основные свойства соединений в газовой фазе являются истинными (собственными) свойствами, и в противоположность случаю в конденсированной фазе практически не обсуждается зависимость этих свойств от строения аминов.
Следует отметить, что, несмотря на большой интерес, проявляемый к результатам по основности аминосоединеиий в газовой фазе, пока еще нет общего подхода к объяснению эффектов их структуры на данное свойство. Выявлены только некоторые закономерности, характеризующие поведение отдельных групп родственных аминов. Например, Тафт рассмотрел изменение основности при переходе от аммиака к первичным, вторичным и третичным аминам и нашел, что введение одной, двух или трех алкильных групп (СН3; С2Н5; n-С3Н7; Н2С=СН—СН2; НСЕºС—СН2) сопровождается ростом величин DGB в соотношении 1,00 : 1,72 : 2,22. Повышающее основность действие метильных групп при последовательном накоплении их в a-положении может быть представлено пропорциональностью 1,00: :1,85 : 2,60 [7]. Введение метильной группы в a-положение увеличивает основность амина примерно на 2,1 ккал/моль, в b-и g-положения — на 0,9 и 0,5 ккал/моль соответственно [155].
При сопоставлении ароматических и алифатических аминов с одинаковым числом углеродных атомов у атома азота было найдено, что изменение гибридизации a-атомов углерода (например, переход от пиридина к N-метилпирролидину, от анилина к циклогексиламину) практически одинаково влияет на изменение основности в воде и газовой фазе [7]. Была также обнаружена приблизительно прямолинейная зависимость между изменениями основности аминосоединений, имеющих одинаковое число углеродных атомов у азота, но разный характер гетероатома, и степенью этой гибридизации [159], а также между основностью алкиламипов и степенью гибридизации b-углеродного атома в алкильном радикале [7]. В тех случаях, когда варьирование заместителя происходит не у реакционного центра, были выявлены более строгие закономерности влияния структуры на основность аминов. Так, найдена корреляция между DGB для a-замещенных триметиламинов и sI этих заместителей [7]. Величины DGB для 3-й 4-замещенных пиридинов хорошо коррелируют [7, 162, 163] с их основностью в воде и с постоянными sI (s°) и sR+ (sG+) характеризующими электронные эффекты заместителей [7, 158, 163]. Аналогичные зависимости (но менее строгие) можно получить и при подобных сопоставлениях основности замещенных анилинов в газовой фазе [3, 7].
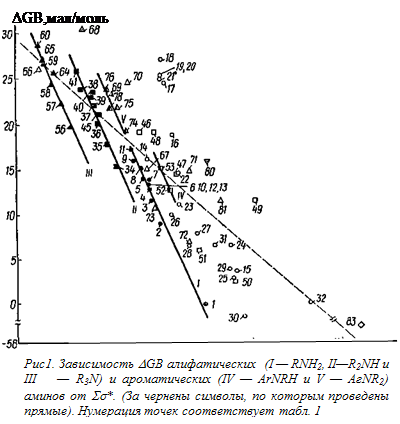 Непосредственное
сравнение величин DGB (см. табл. 1) со
значениями Ss* заместителей*, присоединенных к атому азота, показывает
что, на первый взгляд, здесь отсутствует какая-либо зависимость. Тем не менее
имеется некоторая тенденция к уменьшению основности рассматриваемых соединений
с ростом электро-отрицателыюсти заместителей в них. Это позволило через 40 (из
47) точек для различных аминосоединений (алкиламины, ариламины, производные
гидразина и амиды), основности которых в газовой фазе были известны к концу
1974 г., провести прямую, описываемую [158] уравнением**
Непосредственное
сравнение величин DGB (см. табл. 1) со
значениями Ss* заместителей*, присоединенных к атому азота, показывает
что, на первый взгляд, здесь отсутствует какая-либо зависимость. Тем не менее
имеется некоторая тенденция к уменьшению основности рассматриваемых соединений
с ростом электро-отрицателыюсти заместителей в них. Это позволило через 40 (из
47) точек для различных аминосоединений (алкиламины, ариламины, производные
гидразина и амиды), основности которых в газовой фазе были известны к концу
1974 г., провести прямую, описываемую [158] уравнением**
DGB = (2,1±0,1) — (6,46±0,16)Ss*, (s = 2,1; r = 0,988). (3)
Если аналогичную прямую (пунктирная линия на рис. 4) провести через каждую 71 точку, представленную на указанном рисунке, то ее уравнение имеет вид
DGB = (23,9 ± 0,7) — (8,94 ± 0,48) Ss*,
(s = 4,73; r =0,914). (4)
Следует отметить, что в этом случае при сравнительно узких доверительных интервалах в параметрах уравнения (20) на рис. 4 наблюдаются довольно значительные отклонения от указанной прямой/
Например, точка для аммиака (№ 1) отклоняется вниз, а для тетра-метилендиамина (№ 18) — вверх почти на 11 ккал/моль. Более того, и так невысокий (0,914) коэффициент корреляции значительно уменьшается (до 0,798) при исключении из рассмотрения далеко отстоящей точки для №3 (№ 83). Поэтому найденную зависимость (уравнение (4)), вероятно, можно рассматривать как качественное соотношение, отражающее указанную выше тенденцию к уменьшению DGB с ростом электроноакцепторности заместителей в аминосоединеииях.
Интересные результаты получаются при рассмотрении величин DGB для алкиламинов с насыщенными углеводородными радикалами. Как видно из рис. 4, соответствующие точки (полностью зачерненные символы) группируются таким образом, что для первичных, вторичных и третичных аминов можно провести отдельные прямые [164] с наклонами, соответственно равными: —22,8 ± 2,2; —23,9 ± 2,7 и —23,5 ± 2,2.
Наличие отдельных прямых для алкиламинов различных классов не является неожиданностью. Так, при подобной обработке (сопоставление с Ss*) потенциалов ионизации — одной из важнейших составляющих сродства к протону в газовой фазе [47, 151, 153]) — было найдено [165], что по аналогии с корреляцией потенциалов ионизации различных органических соединений RxМНy (где М =С, О и S) эти данные лучше всего представлять в виде отдельных зависимостей для первичных, вторичных и третичных аминов, хотя имеется и другой подход, в соответствии с которым зависимость потенциалов ионизации аминов от их структуры описывается единым уравнением [166]. Однако первый подход более предпочтителен, поскольку он охватывает больший набор аминов, а также рассматривает с единых позиций потенциалы ионизации самых различных соединений*. Кроме того, при сопоставлении величин РА с потенциалами ионизации [153, 155] и энергиями связывания остовных (1s) электронов [167] наблюдается также отдельные прямые для разных классов аминов. Следует отметить, что при сравнении термодинамических характеристик процессов протонирования аминов в газовой и конденсированной фазах, общей и электростатической теплот гидратации алкиламмоний-ионов как с величинами РА, так и с радиусами этих ионов были получены отдельные прямые для первичных, вторичных и третичных аминов [3, 140, 153]. При этом амины с электроотрицательными заместителями в тех случаях, когда соответствующие данные рассматривались, заметно отклонялись от найденных зависимостей [140]. Из рис. 1 отчетливо видно, что точки (незачерненные символы) для аминов, содержащих электроотрицательные заместители, отклоняются (иногда существенно) от полученных прямых, т.е. здесь наблюдается то же явление, что и при сопоставлении величин DН протонирования аминов в воде и газовой фазе.
Отклонения, наблюдаемые для диаминов (табл. 1, № 16—21), обусловлены внутримолекулярной сольватацией типа III [156,157]. Влияние этой сольватации, которое можно количественно оценить по отклонению соответствующих точек от прямой I на рис. 4, сильнее всего проявляется при n = 4, что можно связать с устойчивостью соответствующих структур.
Внутримолекулярная сольватация того же типа, вероятнее всего, ответственна и за отклонения вверх точек для b-метоксиэтиламина (№ 22), пиперазина (№ 46), морфолина (№ 47) и N,N-тетраметилэтилендиамина (№ 68) от соответствующих прямых. В случае диазобициклооктана (№ 69) существенное отклонение (~ 13 ккал/моль) точки о! прямой для третичных аминов, вероятно, обусловлено стабилизацией его катиона за счет взаимодействия неподеленной электронной пары непротонированного атома азота с орбиталым атомом азота, к которому присоединен протон.
 |
Отклонения точек для аминов, содержащих электроотрицательные заместители, также, по-видимому, следует связывать с увеличением DGB этих аминов за счет стабилизации их катионов при образовании внутримолекулярных водородных связей, например типа IV для фторсодержащих алкиламинов.
Труднее объяснить наблюдаемые отклонения от соответствующих прямых точек для циклогексиламина (№1 4), гидразина (№ 15), манксина (№ 66) и N,N-диметилгидразина (№ 67). Здесь, по-видимому, проявляется как некоторое расхождение в величинах GB, полученных разными авторами (например, в случае манксина приведенное в табл. 1 значение DGB было рассчитано при сопоставлении данных по РА этого амина и GB других аминов), так и влияние (в гидразинах) неподеленной электронной пары на α-гетероатоме (α-эффект ).
При
рассмотрении основности ароматических аминов в газовой фазе (табл. 1, № 31, 52,
53,74—78), прежде всего обращает внимание тот факт, что их величины DGB значительно выше, чем для аммиака, и
практически совпадают с таковыми для алифатических аминов с насыщенными
углеводородными заместителями. Такое аномальное поведение анилина и его
производных объясняется повышенным влиянием поляризуемости фенильного кольца в
газовой фазе, которое превышает действие резонансного эффекта. Указанное
влияние поляризуемости α-непредельных связей проявляется и в случае дифенил-
и трифениламинов. Так, трифениламин, основность которого в воде не поддаётся
измерению в газовой фазе,оказался сильнее, чем даже метиламин. Повышена
основность и дифенилами на, который по своему сродству к протону в газовой
фазе находится между метиламином и анилином. Используя отданные, можно
попытаться количественно оценить различие во влиянии поляризуемости и
резонанса фенильной группы на основность ариламинов. Для анилина, где
соответствующая величина расчитывалась как отклонение его точки от корреляционной
прямой для первичных алкнламинов, она оказалась равной примерно 10 ккал/моль. В
случае дифениламина (отклонение от прямой для вторнчных алкиламинов) при
использовании среднего значения DGB
между анилином и метиламином (~ 8 ккал/моль) получается, что действие
каждой фенильной группы равно ~ 10 ккал/моль. А для трифениламина (DGB = ~ 11 ккал/моль как среднее значение
между метиламином и М-метиланилином данная величина, определенная по
отклонению от прямой для третичных алкиламинов, оказа лась равной ~ 11
ккал/моль. Таким образом, можно считать, что; различие в действии
эффектов поляризуемости и резонанса
каждой α-кратной связи практически не зависит от числа таких связей.
Влияние только резонансного эффекта количественно оценивается при
сравнении основности в газовой фазе бензохинуклиди-1 на (№ 79) и
N,N-диалкиланилинов (№ 74—78). Сопоставление значений DGB для этих аминов приводит к величине ~ 5 ккал/моль, Принимая
во внимание отмеченное выше различие во влиянии поляризационного и
резонансного эффектов фенильных групп, можно считать, что эффект поляризуемости
α-непасыщеной связи на газофазную основность аминов равен ~ 15 ккал/моль.
Вероятно, вследствие проявления эффектов поляризуемости пиррол (№ 54 в газовой фазе из-за повышающего основность влияния двух α-кратных связей оказался основнее аммиака на 4 ккал/моль.
Влияние поляризуемости, по-видимому, является ответственным за значительное повышение основности газовой фазе пиридина (№ 80) амидов* (№ 32, 33, 55, 81, 82) по сравнению с аммиаком и алкил- аминами.
Рассмотренные данные показывают, что влияние поляризуемости непредельных группировок на основность аминов в газовой фазе оказывается весьма эффективным (оно значительно превышает резонансные влияния). В то же время поляризуемость насыщенных радикалов, которая должна увеличивать основность соединения с ростом числа заместителей у реакционного центра в данном случае практически не проявляется, поскольку третичные алкиламины являются более слабыми основаниями, чем вторичные и первичные при равных величинах Σσ*(ср. расположение прямых I - III на рис. 1).
Интересно сопоставить основность в газовой фазе трехфтористого азота (см. № 83 в табл.1) и аммиака (№ 1). Пониженная основность NF3 в первом приближении может быть объяснена акцепторным действием трех атомов фтора у азота. Однако при количественном рассмотрении получается, что с учетом величины Σσ* атомов фтора значение ΔGB для этого соединения должно быть равным примерно — 190 ккал/ моль. Повышение наблюдаемой величины над расчётной (примерно на 130 ккал/моль) трудно объяснить на основе любых известных эффектов атомов фтора. Однако возможно, что здесь протоиирование осуществляется не по атому азота, а по атом у фтора. В пользу этого может свидетельствовать тот факт, что величины РА для НF, СН3F и С2Н5F равны 137, 151, 163 ккал/моль соответственно, т. е. практически совпадают со значением для NF3 (151 ± 10 ккал/моль ).
Следует отметить, что влияние алкильных заместителей у атома азота в анилине оказывается аналогичным таковому для алифатических аминов, т. е. основность их увеличивается с ростом числа и размера радикалов (ср. № 31, 52, 53, 74—78), и это влияние удовлетворительно описывается уравнением типа (1). Из рис. 1 видно, что точки (частично зачерненные символы) для N-алкил- и N,N-Диалкиланилинов ложатся на отдельные прямые практически с тем же наклоном, что и для алифатических аминов.
В связи с тем, что наклоны прямых па рис. 1 для алифатических и ароматических аминов практически совпадают, все рассмотреные данные для 34 аминов были обработаны по единому уравнению. В соответствии с этими расчетами влияние структуры названных аминов описывается следующими уравнениями
DGB = 32,7 ± 0,2 — 23,1 ± 0,З Σσ*
(первичные алкиламины), (5а)
DGB = 27,6 ± 0,3 — 23,1 ± 0,3
Σσ* (вторичные алкиламины), (56)
DGB = 20,3 ± 0,3 — 23,1 ± 0,3
Σσ* (третичные алкиламины), (5в)
DGВ = 38 ± 0,5 — 23,1 ± 0,3
Σσ* (N-алкиланилины), (5 г)
DGB = 32,6 ± 0,4 — 23,1 ± 0,З Σσ* (N,N-диалкиланилины), (5д)
(s-0,731, R = 0,990).
При этом оказалось, что первичные алифатические и третичные ароматические амины случайно ложатся практически на одну и ту же линию (прямые I и V на рис. 1). Величина ρ* (~ — 17, если перевести ее в размерность рКа) здесь оказалась значительно выше, чем для воды (~-3) и других заместителей.
Расположение прямых на рис. 4 свидетельствует о том, что в газовой фазе сродство аминов к протону при равенстве Σσ* их радикалов изменяется в ряду: первичные> вторичные> третичные
В+Н · (Н2О)п-1 + Н2О « В+Н • (Н20)n (6)
Эти данные свидетельствуют о том, что, например, кластер |МН4 (Н20)4 практически не обладает особой устойчивостью по сражению с кластерами другого состава, поскольку на графиках «свойство — n» (n изменяется от 1 до 5) некоторый излом при N = 4 обнаруживается только при рассмотрении изменений энтальпии процесса В случае изменений свободной энергии [180] никакого излома не наблюдается, хотя при преимущественном образовании первого гидратного слоя в соответствии с рассмотренной, выше сольватационной теорией следовало бы ожидать различный характер обеих указанных зависимостей в области n < 4 (образование первого гидратного слоя) и n > 4 (образование следующего слоя), т. е заметные изломы при n = 4 Для катиона триметиламмония соответствующий график как для DH°, так и для DG° не претерпевает никаких изменений при любых n (от 1 до 5) Аналогичная монотонная зависимость соблюдается при любых n [от 1 до 8) при гидратации протона в газовой фазе.
Таким образом, рассмотрение закономерностей влияния структуры аминов на их основность в газовой фазе показало, что эго влияние оказалось не проще, чем в конденсированной, а даже несколько сложнее, поскольку здесь наряду с эффектами, действующими в растворах, проявляются и другие факторы. Поэтому использование величин GB или РА, являющихся, по мнению Арнетта, “наиболее подходящей характеристикой основности”, для оценки влияния строения аминосоединений на их свойства и эффектов сольватации из-за сложности учета всех указанных факторов в настоящее время пока затруднительно. Тем не менее выявленные закономерности дают основание полагать, что с накоплением нового экспериментального материала положение в этой области существенно прояснится.
Удалось показать, что значения РА подчиняются принципу линейности свободных энергий (ЛСЭ). Так, при изменении свойств заместителя в g-положении пиридинового цикла наблюдаются хорошо коррелирующие зависимости с электронными константами si и sr + (10)
РА/РА0 = 1б,7s I + 10 , 3 sr + (8)
где РА° и РА - сродство к протону пиридина и пиридина, замещенного в g -положении. Интересно, что основность пиридинов в газовой фазе в большей степени чувствительна к электронным эффектам заместителей, чем в водном растворе. Более подробно причины этих различий, которые часто принимают характер инверсии, будут обсуждаться дальше. Здесь же отметим, что основность соединения в соответствии со схемой (4) определяется разностью свободных энергий нейтральных молекул и соответствующих протежированных форм. Бри этом главный вклад в энергетический баланс вносит стабильность ионов BН+.
Ионы в газовой фазе не стабильны. Раз образовавшись, они быстро гибнут в результате рекомбинации с ионами противоположного знака или на стенке [5]. Наиболее неустойчивы в газовой фазе простейшие ионы Н30+, NН4+ . В жидкой среде ионы стабилизируются за счет сольватации, энергия которой может превысить энергию образования иона из молекулы. В этом случае можно ожидать инверсии основности при сопоставлении данных в газовой фазе и в растворе. Строение многих слабых органических оснований способствует делокализации образовавшегося при протонировании заряда. Такие ионы стабильны в газовой фазе, а основность соответствующих оснований при прочих равных условиях будет выше. Именно возможностью делокализации положительного заряда в анилиниевом ионе объясняется более высокая по сравнению с аммиаком основность анилина в газовой фазе, тогда как в воде аммиак - значительно более сильное основание, чем анилин [5,11].
Таким образом, эксперименты в газовой фазе позволяют выдвинуть критерий сравнения основности соединений (в том числе и слабых оснований), проследить влияние на основность заместителей в реакционных сериях. Во всех подобных случаях в качестве такого критерия рассматривается сродство к протону РА. Однако экспериментальная техника определения РА пока еще чрезвычайно сложна и недоступна для большинства химических лабораторий. Кроме того, в сложных случаях, когда возможно присоединение протона к более чем одному центру основности соединения (а такие ситуации - отнюдь не редкость), интерпретация полученных экспериментально параметров вызывает существенные затруднения. 3 связи с этим были предприняты попытки установления линейных зависимостей между РА и другими параметрами, более доступными экспериментально и адекватно отражающими сложный характер протонирования.
В работах [12, 13] одновременно и независимо предложена линейная зависимость между РА и энергией ионизации для кислород- и азотсодержащих оснований:
РА = - IЕ(Х1s)
+ const (9)
где IЕ(Х1s) - энергия ионизации
для 1s-электрона центра основности - атома кислорода (X = 0) или азота X = М).
Теоретическое обоснование зависимости (9) базируется на сходстве моделей
протонизации и удаления 1s-электрона при ионизации, И в
том и в другом случае энергия может быть выражена суммой двух членов (термов):
один терм связан с электронной плотностью на орбитали, ответственной за
ионизацию (initial state),
другой - со стабилизацией заряда после ионизации (final
state).Заместители могут оказывать влияние как на
первый терм (индуктивный эффект), так и на второй (поляризационный эффект),
причем последний в случав органических оснований обычно преобладает в обеих
моделях ионизации [13]. Вместо IЕ в уравнении (9) было предложено использовать
потенциал ионизации (IР) [13,14]. По мнению авторов, применение величины IР*
более корректно в случае сложных органических молекул, направление протонизации
в которых может быть неоднозначным. В работе [13] приведены корреляционные
соотношения между РА и IР и, в частности, для карбонильных соединений (РА =
-0,690 IР + 15,03) и для спиртов и эфиров (РА = - 0,397 IР + 12,29). Подводя
итог рассмотрению экспериментальных исследований процесса переноса протона в
газовой фазе, отметим, что найденные в этих работах критерии полезны и
пригодны для количественного сравнения основности в данных условиях*. Более
того, из сопоставления экспериментальных данных, полученных для одних и тех же
соединений в газовой фазе и в растворе, могут быть выполнены количественные
оценки энергии сольватации про тонированных оснований. В основу таких расчетов
положен термодинамический цикл Борна [б].
Однако довольно часто при изучении протонирования органических оснований возникает необходимость установить центр присоединения протона. В тех случаях, когда органическая молекула обладает несколькими вероятными центрами основности, не всегда можно предугадать, куда именно присоединится протон. При этом весьма желательно иметь наглядное представление об изменении электронной структуры и конфигурации молекулы в результате протонирования. Как правило, экспериментальные метода не дают однозначного ответа на эти вопросы. Кроме того, некоторые зависимости, установленные экспериментально, например (8), (9), нуждаются в теоретической интерпретации. В этих случаях на помощь приходят квантово-химические методы исследования.
3. Закономерности, выявленные для основности
Наличие свободной электронной пары у атома N придаёт аминогрупам основные свойства. В соответствии с теорией Брауна. Основание является акцептором протона: образуется протонированная форма амина. Согласно определению Льюиса, атом может образовывать связь с кислотами Льюиса, т. е. С любыми частицами, имеющими орбиталь, способную принять участие в создании связи с использованием электронной пары основания.
![]() R3N: + AH R3N+H
+ A-
(1)
R3N: + AH R3N+H
+ A-
(1)
Равновесие кислота — основание устанавливается довольно быстро, и при обсуждении основности следует рассмотреть положение равновесия в данной системе. Оно определяется разностью свободных энергий DG0 основания и сопряженной кислоты. На относительную устойчивость этих двух частиц влияют три основных фактора: электронные факторы, природа растворителя и структурные особенности, которые будут рассмотрены ниже.
Влияние электронных факторов на основность можно оценить с помощью данных об основности в газовой фазе, полученных рядом методов, таких, например, как масс-спектрометрия или ионный циклотронный резонанс. Эти методы позволяют изучать ион-молекулярные взаимодействия и рассчитывать DG0 по уравнению 3, где GB — основность в газовой фазе,
![]() АН+В ВН + А (2)
АН+В ВН + А (2)
DG0 = GB(А)-GB(В)
(3)
Результаты, полученные при изучении большого числа алкиламинов, были использованы для количественной оценки влияния алкильных групп на основность самих аминов. Порядок возрастания этого влияния согласуется с увеличением электронодонорного «индуктивного эффекта» алкильных групп, который оказывает сравнительно более сильное стабилизующее воздействие на протонированные формы аминов, чем на свободные амины. В соответствии с этим наблюдается увеличение основности, например, в следующем ряду:
NН3 < МеNН2 < ЕtNН2 < н-BuNН2 < Ме2NН < Ме3N < Еt3N < н-Вu3N
Понятие “индуктивный эффект” достаточно хорошо обосновано экспериментально и является очень полезным для химиков-органиков, однако стало очевидным, что алкильные группы способны стабилизовать не только положительные, но и отрицательные ионы. Это следует из характера кислотности спиртов в газовой фазе (ВuОН > EtOН > МеОН > Н2О) и влияния алкильных заместителей на увеличение силы кислоты в растворе. Этот эффект можно представить как результат делокализации .положительного или отрицательного заряда в молекуле вследствие поляризации различных связей. Как и следует ожидать, .электроотрицательные атомы понижают основность, например, в ряду:
СН3СН2NН2 > FСН2СН2NH2 > F2СНСН2NН2 > F3ССН2NH2
Оценка основности в газовой фазе относительно аммиака
![]() NH4 + B NH3 + BH
NH4 + B NH3 + BH
DG0 = GB(NH3) - GB(В)
Таблица 2. Основность некоторых аминов в газовой фазе
| соединение |
DG0, кДж/моль |
соединение |
DG0, кДж/моль |
|
|
-101,7 |
|
-75,0 |
|
|
-103,0 |
|
-46,9 |
|
|
-84,2 |
|
-64,9 |
|
|
-84,6 |
|
-28,1 |
|
|
-75,4 |
|
-47,3 |
|
|
-54,4 |






дала возможность сравнить циклические и ациклические амины (табл. 1); важно отметить, что для аминов, имеющих аналогичные заместители, значения DG0 близки (отрицательные значения указывают на большую силу основания). Отклонение, наблюдаемое в случае азиридина, находит объяснение в рамках теории гибридизации: орбиталь свободной электронной пары в азиридине носит более выраженный s-характер, чем в диметиламине, и поэтому менее вероятно ее участие в образовании связи с протоном. Для бензиламинов, по-видимому, экспериментальных данных недостаточно; для трех первичных аминов, представленных в табл. 1, наблюдается удовлетворительная корреляция между характером гибридизации b-углеродного атома и значением DG0 .
Закономерности, выявленные для основности различных аминов в газовой фазе, привлекают своей простотой и четкостью. Большинство экспериментов в органической химии осуществляется в растворах, и в этих случаях изменение основности может иногда описываться приблизительно такими же закономерностями, как и в случае газовой фазы (например, Bu3 > Вu2NН > ВuNН2, полученный для растворов в хлорбензоле относительно 2,4-динитрофенола). Однако часто эти выводы не носят общего характера; так, в бензоле наблюдается следующий порядок основности: Вu2NН > Вu3Н > ВuNH2. В течение многих лет химики проявляли особый интерес к закономерностям, существующим в водных растворах. В этом случае основным параметром является свободная энергия протонирования основания в воде DG0 (Н2О), выражаемая обычно как рKа сопряженной кислоты +ВН [DG0 (Н2О) = —RТlnКа] - Значения рKa для простейших аминов приведены в табл. 2.
Таблица 2. Значение рКа кислот, сопряжённых с алкиаминами (Н2О, 25 0С)
| Соединение |
рКа |
|
| R=Et | R=Me | |
|
R3N |
10,85 | 9,80 |
|
R2NH |
11,09 | 10,73 |
|
RNH2 |
10,80 | 10,66 |
|
NH3 |
9,25 | |
Отсутствие четкой закономерности в поведении алкиламинов, объясняли по-разному. Влияние пространственных факторов на стадии протонирования можно не учитывать, и долгое время признавалась важность эффектов сольватации, протекающей в различной степени. Недавно Ауэ применил эти данные в сочетании с известными термодинамическими параметрами в водных растворах для всестороннего анализа дифференциальной сольватации [142]. В ряду алкиламинов теплоты гидратации обычно закономерно понижаются с увеличением размеров молекул. Это влияние алкильных заместителей, называемое гидрофобными эффектами, изучено недостаточно, однако предполагают, что подобные эффекты почти полностью отсутствуют в нейтральных и протонированных аминах, находящихся в водной системе. Считают, что в растворе важным фактором является влияние на сопряженную кислоту ослабления взаимодействия между растворителем и протонированным амином при делокализации заряда в ионе. К тем же выводам приходят при интерпретации этого явления с точки зрения электростатической сольватации (считают, что энергия сольватации и ионный объем связаны обратной зависимостью) и сольватации с участием специфических водородных связей (при этом каждая специфическая водородная связь ослабляется вследствие делокализации положительного заряда в ионе). Таким образом, в тех случаях, когда усиление поляризуемости вследствие увеличения числа алкильных заместителей приводит к стабилизации иона аммония за счет делокализации заряда, сольватация иона должна происходить менее экзотермично, способствуя ослаблению стабилизующего влияния заместителей по сравнению с тем, что имеет место в газовой фазе. Поэтому в ряду алифатических аминов суммарное влияние увеличения степени алкилирования постепенно ослабевает и может фактически приводить к обращению ряда в тех случаях (например, Ме3N в табл. 2), когда эффект уменьшения стабилизации при сольватации сильнее, чем внутримолекулярное стабилизующее влияние алкильных заместителей. И, наоборот, в тех случаях, когда индуктивные эффекты могут вызывать дестабилизацию иона аммония, ион будет обладать повышенной плотностью заряда на атоме азота и лучше сольватироваться; здесь вновь наблюдается противодействие электронным эффектам. Важность сольватации можно подчеркнуть тем, что изменение свободной энергии при переходе ионов аммония из газовой фазы в водный раствор может составлять до 25— 110 кДж/моль (примерно аналогично изменению DG0 за счет электронных эффектов алкильных заместителей в газовой фазе). Для более подробного и систематического знакомства с термодинамическим аспектом данной проблемы и уяснения природы эффектов сольватации читателю следует обратиться к работам [140—142].
В данном разделе не будет дублироваться обсуждение явления постепенного понижения основности при переходе от алкиламинов к аридаминам и амидам. Понижение основности по мере усиления s-характера азота (например, в пиридине и нитрилах) также освещается в соответствующих разделах.
Как уже отмечалось, данные, говорящие о каком-либо влиянии пространственных факторов на перенос протона в газовой фазе, отсутствуют, но при описании взаимодействия аминов с кислотами Льюиса (уравнение 4) становится важным учет объема заместителей. Этот фактор впервые был рассмотрен в классическом исследовании Брауна:
![]() R3N + BMe3 R3N ® BМe3
(4)
R3N + BMe3 R3N ® BМe3
(4)
Сравнение теплот диссоциации комплексов аминов с кислотами Льюиса и теплот диссоциации соответствующих аммониевых ионов позволяет достаточно точно оценить энергию пространственного напряжения, которое наблюдается у аминов, содержащих заместители различного объема (табл. 3).
Таблица 3. Энергия пространственного напряжения для аддуктов и триметилбора.
| Амин | Энергия пространственного напряжения | Амин | Энергия пространственного напряжения |
|
Me2NH |
5,9 |
Et2NH |
30,1 |
|
|
12,1 |
Трет-BuNH2 |
33,5 |
|
|
18,1 |
Et3N |
71,2 |
|
|
29,3 | ||

Измерение констант равновесия дает сведения о собственной основности ряда аминов, для которых пространственные факторы не меняются, а изменение заместителей происходит на большем удалении от атома азота. Были изучены многие другие комплексы аминов, например комплексы с ионами металлов, комплексы с лантаноидными элементами); с галогенами и полинитросоединениями, включая пикриновую кислоту. Реакция образования пикратов лежит в основе классического метода идентификации аминов.
Аминогруппы обладают способностью к внутри- и межмолекулярной ассоциации друг с другом или с другими функциональными группами. Оба возможных типа водородной связи (амин выступает как донор водорода или как акцептор) иллюстрированы формулами (5) и (6). Термином “водородная связь” в каждом случае принято обозначать более слабую из двух связей с водородом.
![]()
(5)
![]()
(6)
Образование водородных связей происходит в твердом состоянии, в жидкой фазе, в растворе, а иногда даже и в газовой фазе. По прочности водородная связь (~8— 40 кДж/моль) является промежуточной между ковалентными и ван-дер-ваальсовыми связями Особая важность этого типа связи была продемонстрировала в ходе обсуждения основности в водном растворе. Определенное влияние водородных связей на физические свойства выражается в том, что температуры кипения первичных аминов выше, чем температуры кипения углеводородов приблизительно той же молекулярной массы, хотя в случае третичных аминов этот эффект, естественно, исчезает. Были изучены спектроскопические проявления водородной связи; эти наблюдения лежат в основе способов ее обнаружения и изучения. Проблеме водородной связи посвящены краткие обзоры [143] и обширные монографии [144].
* для равновесия в возможна более сложная картина, предусмотренная схемой (5)
* К сожалению, на рис. 1 не нанесён ряд точек из-за отсутствия или неточности, на наш взгляд, величин s* для заместителей в некоторых аминах.
** В качестве стандартного основания был выбран триметиламин, а величина r* дана в ед. рКа.
* Найденные в работах [165, 166] корреляционные зависимости качественно не изменяются, если вместо рассмотренных в них адиабатических потенциалов ионизации воспользоваться данными по вертикальным потенциалам ионизации [155].
* Сравнение величин DGB для амидов и аминов основано на допущении, что амиды в газовой фазе протонируются по атому азота. Однако этот вопрос, как и в случае водных растворов, (см. выше) пока ещё не решён.
* Значения IР могут быть определены экспериментально с помощью метода фотоэлектронной спектроскопии или вычислены путём квантово-химических расчётов.