
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
Психология и педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Рефераты по сексологии
Рефераты по информатике программированию
Краткое содержание произведений
Реферат: Синтез твердых растворов и исследования низкотемпературных фазовых превращений
Реферат: Синтез твердых растворов и исследования низкотемпературных фазовых превращений
МОСКОВСКИЙ ОРДЕНА ЛЕНИНА, ОРДЕНА ОКТЯБРЬСКОЙ РЕВОЛЮЦИИ И ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ имени М.В.ЛОМОНОСОВА
ХИМИЧЕСКИЙ ФАКУЛЬТЕТ
Кафедра неорганической химии
Лаборатория неорганического материаловедения
КУРСОВАЯ РАБОТА
по неорганической химии
студента 104 группы
Орловой Натальи Викторовны
СИНТЕЗ ТВЕРДЫХ РАСТВОРОВ В СИСТЕМЕ Bi2O3-CaO И ИССЛЕДОВАНИЯ НИЗКОТЕМПЕРАТУРНЫХ ФАЗОВЫХ ПРЕВРАЩЕНИЙ
Научный руководитель : младший научный сотрудник Кнотько А.В.
Преподаватель: к.х.н., старший преподаватель Куприянова Г.Н.
Москва - 1997
Содержание.
1. Введение……………………………………………………………………………
2. Литературный обзор……………………………………………………………….
2.1. Фазовые соотношения в системе Bi2O3-CaO…………………………..
2.2. Методы синтеза керамических материалов…………………………….
2.2.1. Керамический метод……………………………………………
2.2.2. Методы химического осаждения………………………………
2.2.3. Золь-гель метод…………………………………………………
2.2.4. Распылительная сушка………………………………………….
2.2.5. Криохимический метод………………………………………
2.3. Фазовые превращения в твердом состоянии…………………………
3. Экспериментальная часть…………………………………………………………
3.1. Синтез твердых растворов Bi2O3-CaO…………………………………..
3.2. Эксперименты по низкотемпературному распаду твердых растворов..
3.3. Методы исследования…………………………………………………….
4. Обсуждение результатов……………………………………………………………
5. Выводы………………………………………………………………………………
6. Литература…………………………………………………………………………..
1.Введение.
В последнее время наблюдается усиленный интерес к исследованиям твердофазных процессов с применением ионных электролитов. Для этих исследований имеет большое значение создание твердых электролитов, работающих в широком температурном интервале. Известны твердые электролиты, работающие при высоких температурах (более 1000 К) (материалы на основе оксидов циркония ZrO2, тория ThO2 и т.д.). В сравнительно низкой температурной области (около 700К) хорошо известны твердые электролиты на основе оксида висмута (Bi2O3), легированного оксидами редкоземельных элементов (РЗЭ) и оксидами щелочно-земельных элементов (ЩЗЭ). Образующиеся твердые растворы имеют низкотемпературную границу стабильности, ниже которой термодинамически устойчивы соединения, не обладающие заметной ионной проводимостью. Поэтому для исследований низкотемпературных фазовых превращений наиболее удобно использовать твердые растворы на основе оксидов висмута, что и являлось главной задачей работы.
Данная работа выполнена в лаборатории неорганического материаловедения кафедры неорганической химии Химического факультета МГУ в рамках проекта РФФИ № 96-03-33097а.
2.Литературный обзор.
2.1. Фазовые соотношения в системе Bi2O3-CaO.
Фазовые равновесия в псевдобинарных системах Bi2O3-MO (M=Ca, Sr, Ba ) изучались многими исследователями [1, 2, 3, 4]. Фазовая диаграмма системы Bi2O3-CaO приведена на рис.1. Такахаши и др.[2] также исследовали электрические характеристики в твердых растворах этой системы . Конфлант и др.[3] представили ее в виде четырех инконгруэнтно плавящихся соединений (Bi14Ca5O26, Bi2CaO4, Bi10Ca7O22 и Bi6Ca7O16) и твердых растворов (два кубических и два ромбоэдрических). Также они обнаружили, что ромбоэдрический твердый раствор стабилизированного кальцием оксида висмута (Bi2O3) был изоструктурен системе оксидов Bi2O3-CdO (ранее это было исследовано Силленом [5]). Такахаши и др. [2] нашли, что ромбоэдрическая фаза показывает высокую кислородную и ионную проводимость. При этом при понижении температуры ниже 690С кубический твердый раствор претерпевает эвтектоидный распад на моноклинный оксид висмута (растворимость кальция в котором пренебрежимо мала) и ромбоэдрический твердый раствор, который в свою очередь при температуре 725С переходит в ромбоэдрический раствор другой структуры. Выше 840С и до температуры плавления существует только кубический твердый раствор.
2.2. Методы синтеза керамических материалов.
2.2.1 Керамический метод.
В большинстве случаев для получения оксидных материалов активно используется так называемый “керамический метод”, заключающийся в тщательном механическом смешении оксидов и повторяющихся циклов “обжиг-помол” для полного обеспечения твердофазного взаимодействия. В ряде случаев вместо оксидов используется более легкодоступные карбонаты, нитраты или другие соли.
Рис. 1. Фазовая диаграмма системы CaO - 1/2 Bi2O3 [4].
Этот метод достаточно традиционен при получении любых видов конструкционной и функциональной керамики, однако он обладает рядом существенных недостатков. Главный его недостаток - длительность термической обработки из-за достаточно крупной кристалличности и неоднородности смешения реагентов. При этом чаще всего имеет место неконтролируемый рост кристаллов и, как следствие, помимо химической еще и гранулометрическая неоднородность без того анизотропных зерен керамических материалов, приводящая к невоспроизводимости электрических и магнитных свойств.
Поэтому большое число исследований в области технологии керамических материалов связано с разработкой и применением так называемых “химических методов” получения порошков. Химические методы позволяют повысить гомогенность продукта за счет смешения компонентов в растворе на молекулярном уровне и сохранения этого уровня (с большими или меньшими успехами) на последних стадиях. Получаемые порошки имеют достаточно высокую удельную поверхность и, как следствие, активны в процессах твердофазного взаимодействия. Ниже будут рассмотрены наиболее распространенные “химические методы” получения керамических образцов [6,7].
2.2.2. Методы химического осаждения.
Методы химического осаждения заключаются в совместном осаждении компонентов керамики из раствора в виде нерастворимых солей. Эти методы получили широкое распространение для синтеза разнообразных видов керамики. При правильной постановке эксперимента в ряде случаев удается воспроизводимо получить гомогенную дисперсную смесь солей с заданным соотношением катионов. В идеале оптимальным можно считать такие условия (нереализуемые, в полной мере, на практике из-за различия химических свойств компонентов), когда катионы из раствора осаждаются одновременно и с одинаковой скоростью. Наиболее распространены два типа химического осаждения - то оксалатный и карбонатный методы.
-для осаждения оксалатов в качестве исходных реагентов используются нитраты или ацетаты , а в качестве осадителей - смеси щавелевая кислота - аммиак, щавелевая кислота - триэтиламин , либо избыток насышенного раствора оксалата аммония при фиксированной кислотности раствора, либо водный раствор диметилоксалата . Процесс осаждения осложняется сильной и неодинаковой зависимостью растворимости оксалатов кальция и висмута от величины рН и от концентрации исходных реагентов.
-принципиально осаждение карбонатов аналогично оксалатам. В качестве осадителя при осаждении карбонатных солей используется избыток гидрокарбоната аммония, избыток карбоната натрия, карбонат тетраметиламмония. Осаждение проводят при рН>8, создаваемым добавлением раствора аммиака или гидроксида натрия. В последнем случае, равно как и в случае осаждения карбонатом натрия, приходится уделять особое внимание стадии промывки осадка, поскольку примеси щелочных металлов могут изменять свойства получаемых материалов ( этот процесс может сопровождаться селективным растворением и нарушением стехиометрии).
2.2.3. Золь-гель метод.
Этот метод основан на способности хелатных комплексов с ионами металлов (например цитратных) образовывать при нагревании (100-140С) с многофункциональными спиртами (например, этиленгликолем) низкомолекулярных олигомеров (этерификация) (рис.2). При нагревании последних происходит дальнейшая полимеризация и образуется вязкая смола (гель), при разложении которой получается оксидный порошок.
Если предположить, что комплексы керамикообразующих металлов гомогенно распределены в растворе и это распределение сохраняется и после полимеризации, то не существует причин, из-за которых нарушается гомогенность при разложении. Это обстоятельство позволяет рекомендовать золь-гель метод для работ, направленных на выявление примесей или замещений на свойства керамических материалов.
Рис.2. Схема процессов, происходящих при цитратном золь-гель синтезе.
Метод сам по себе не дорог, т.к. практически не требует аппаратуры (отсутствие операции центрифугирования, фильтрации, промывки и сушки), а в качестве исходных материалов чаще всего используются доступные нитраты.
Препаративно цитратный вариант золь-гель процесса осуществляется следующим образом [8]. В смеси водного раствора нитратов и этиленгликоля (иногда добавляют аммиак для повышения рН до 3-5) добавляют раствор лимонной кислоты в соотношении 1 г-эквивалент кислоты на 1 г-эквивалент металла. Этиленгликоль обычно берут в избытке, поскольку гидроксильные группы стабилизируют в растворе металл-цитратные комплексы и способствуют образованию низкомолекулярных олигомеров.
Другой метод, часто относимый к числу “золь-гель” процессов - так называемая алкоксотехнология. Она основана на получении порошков (или тонких пленок) при медленном гидролизе смеси растворов алкоголятов металлов. Метод перспективен для получения небольших количеств очень чистых и гомогенных порошков, а также волокон, пленок, керамики.
Недостаток метода - малая доступность и дороговизна исходных для синтеза реактивов. Кроме того, специфическая для керамических материалов проблема алкоксидного метода заключается в трудности приготовления гомогенной смеси алкоксидов, поскольку практически не существует алкоголятов кальция, растворимых в распространенных растворителях.
2.2.4. Распылительная сушка.
Ни один из химических методов получения керамических материалов не получил такого распространения, как метод распылительной сушки. Это наиболее крупномасштабный путь получения мелкодисперсных, активных порошков для производства керамических материалов. Суть метода состоит в том, что смесь растворов солей, переведенная посредством ультразвукового распылителя в состояние аэрозоля с размером частиц 0.5-0.8 мкм, переносится газом-носителем в горячую камеру, где происходит мгновенное (полное или частичное) разложение частиц; образовавшийся оксидно-солевой продукт собирают на фильтре.
Смешение компонентов в растворе на атомном уровне, практически мгновенное обезвоживание и разложение микрокапель аэрозоля позволяет получить гомогенный продукт, избежав характерные керамическому методу процессы повторного перемола и обжига, загрязняющие продукт и приводящие к ненормированному росту зерен. Вместе с тем получаемые порошки могут загрязняться материалами, из которых сделана камера для распыления (высокие температуры, присутствие свободной кислоты); помимо этого для того, чтобы избежать образование карбонатов, приходится тщательно очищать большие объемы газа-носителя (кислорода) от примесей СО2.
Наиболее распространенным для данного метода типом солей являются нитраты.
2.2.5.Криохимический метод.
Недостаток большинства химических методов синтеза керамических порошков удается в значительной мере устранить при их синтезе методом криохимической технологии [9]. Суть ее сводится к получению тонкодисперсного и высокогомогенного солевого раствора (а затем и оксидного) прекурсора посредством быстрого замораживания тонко распыленного раствора солей (получение криогранулята) и последующего сублимационного удаления воды. При этом необходимо стараться проводить эксперимент в условиях, исключающих протекания физико-химических процессов, приводящих к нарушению химической и гранулометрической однородности продукта. Таковыми могут быть:
1.Расслоение распыляемых микрокапель на области обогащенные и обедненные растворителем из-за недостаточно высоких скоростей охлаждения (этого удается избежать, если распылять раствор не в жидкий азот (или охлажденный гексан), а на массивную охлажденную до температуры жидкого азота металлическую пластину).
2.Подплавление криогранулята в процессе сублимационной сушки за счет образования низкоплавких эвтектик (в случае растворов нитратов - эвтектики H2O-HNO3 с температурой -43С). Для этого пытаются заменить, где возможно, растворы нитратов на ацетатные (но тогда возникает проблема образования и разложения карбонатов), или нитрат-нитритных, либо используют разбавленные растворы нитратов с невысокой кислотностью (0.1 моль по висмуту с рН 0.7).
3.Сегрегация компонентов продукта сублимационной сушки (содержащего еще до 3 масс.% воды) при его термической обработке за счет плавления гидратов Ca(NO3)2*4H2O (42C) и Ca(NO3)2*3H2O (112C). В этом случае сушку рекомендуется проводить в тонком слое при медленном (5С/час) нагревании до 125С в токе аргона.
4.Нежелательное образование карбонатов из-за присутствия СО2 в атмосфере при отжиге. Недостаточно использовать в этом случае очищенный от углекислого газа воздух - более эффективно создание необходимого парциального давления кислорода смешением кислорода и аргона в соответствующих количествах.
Использование мелкодисперсных (сотни ангстрем) и высокогомогенных прекурсоров, приготовленных с использованием приемов криохимической технологии позволило получить трудно синтезируемые с использованием других методов керамические фазы.
2.3. Фазовые превращения в твердом состоянии.
При последовательном построении теории фазовых превращений [10] рассматриваются причины изменения фазового состояния и механизм превращения. Причиной фазовых превращений является изменение стабильности фаз в зависимости от внешних воздействий. Например, стабильная в определенной температурной области фаза становится нестабильной при понижении или повышении температуры.
При любом фазовом превращении в твердом состоянии происходит перестройка атомной структуры системы. В твердом состоянии перестройка структуры имеет место, кроме того, при процессах, не являющимися фазовыми превращениями, например, при рекристаллизации, пластической деформации скольжением и двойникованием. Такие процессы отличаются от фазовых превращений причиной перестройки кристаллической решетки: атомы занимают новые положения под действием поверхностных или упругих сил, внешнего напряжения, а не вследствие того, что термодинамический потенциал одной конфигурации атомов ниже, чем другой.
Классификация фазовых превращений может быть проведена на основе сравнения фазового состава системы в начальном и конечном состояниях. Продукт фазового превращения может отличаться от исходной (матричной) фазы:
1. составом при сохранении координации атомов в решетке (изоструктурный распад твердого раствора);
2. структурой и фазовым составом (эвтектоидный распад, выделение избыточной фазы);
3.кристаллической структурой, т.е. координацией атомов в решетке (мартенситное и массивное превращения, упорядочения атомно-кристаллической структуры).
Другая классификация фазовых превращений в твердом теле может быть основана на механизме протекающих процессов.
Диффузионные процессы. К ним относятся как процессы, протекающие через стадии образования и роста зародышей новой фазы, так и процессы, связанные с расслаиванием твердого раствора на участки с большей и меньшей концентрацией компонента (спинодальный распад, образование зон Гинье - Престона).
Бездиффузионные процессы. Это процессы, происходящие за счет коллективных перегруппировок атомов на расстояния порядка межатомных (например мартенситное и массивное превращения).
Вывод о протекании процесса по тому или иному механизму можно сделать исходя из морфологии полученного продукта по данным электронной или оптической микроскопии.
3. Экспериментальная часть.
3.1. Синтез твердых растворов Bi2O3-CaO.
Для первой стадии эксперимента в качестве исходных веществ были взяты оксид висмута (Bi2O3) и карбонат кальция (CaCO3) в мольных соотношениях Bi2O3/CaCO3 - 0.96:0.04, 0.9:0.1, 0.8:0.2, 0.76:0.24, 0.7:0.3, 0.6:0.4, 0.5:0.5. Квалификация всех исходных препаратов была не ниже “ч.д.а.”. Реагенты смешивались для получения как можно более однородной смеси, после чего прокаливались на воздухе при температуре 750С в течение 24 часов в трубчатой печи с последующим рентгенофазовым анализом полученных образцов. В ходе реакции
0.5(1-х) Bi2O3 + х CaCO3 == Bi1-хСахО1.5-0.5x+ х CO2 (спекание при 750С, 24 часа)
выделялся углекислый газ (CO2) и образовывался желтый порошок. Для неоднофазных, по данным РФА , образцов цикл “помол-отжиг” был повторен при тех же условиях.
Полученный в результате предыдущей стадии эксперимента порошок состава Bi1-xCaxOy (х = 0.02, 0.05) был растворен в разбавленном растворе азотной кислоты (30%) и упарен на горелке до начала выделения BiONO3 в виде тонкой пленки на поверхности раствора:
Bi(NO3)3 = BiONO3 + 2 NO2 + 0.5 O2
После этого был произведен так называемый “бумажный синтез” [11] : образовавшимся раствором пропитали несколько листов бумажного фильтра (для предотвращения расслоения компонентов раствора при разложении) и производили разложение на горелке до окончания реакции восстановления избыточной азотной кислоты глюкозой, образовавшейся при гидролизе целлюлозы бумаги (признаком окончания реакции было прекращение выделения бурого газа NO2), нитрат висмута при этом полностью разложился до оксинитрата. Затем продукт реакции в течении 30 минут отжигали в муфельной печи при температуре 750С, а после произвели закалку на воздухе. При этом происходило разложение нитратов с одновременным образованием твердого раствора:
(1-х) BiONO3 + x Ca(NO3)2 = Bi1-xCaxO1.5-0.5x + (1+2x) NO2 + 0.25(1+2x) O2
Следующей стадией эксперимента был помол полученного вещества с последующей обработкой в трубчатой печи в течение 24 часов при температуре 750С, с последующей закалкой на воздухе и снятием рентгенограмм полученного вещества.
3.2. Эксперименты по низкотемпературному распаду полученного вещества.
Для исследования низкотемпературного распада твердого раствора Bi0.98Ca0.02O1.49 была проведена серия отжигов при температуре 550С на воздухе с временами 1, 2, 5, 15 минут и последующим рентгенографическим и электронномикроскопическим анализом полученных образцов.
3.3. Методы исследования.
Для исследования полученных образцов были использованы методы рентгенофазового анализа и просвечивающей электронной микроскопии.
Для рентгенофазового анализа образцы снимали на дифрактометре ДРОН-3М (излучение CuKaср) с шагом съемки 0.1 О, временем экспозиции 2 сек., интервалом съемки 20О-60О, в качестве внутреннего стандарта для расчета параметров элементарной ячейки использовали кремний.
Полученные рентгенограммы обрабатывали при помощи пакета программ “POWDER” (лаборатория направленного неорганического синтеза, Химический факультет МГУ) и оригинальных программ, разработанных в лаборатории неорганического материаловедения Химического факультета МГУ.
Обработка данных, полученных на дифрактометре, включала в себя следующее:
1. вычисление межплоскостных расстояний;
2. сравнение полученной рентгенограммы с данными библиотеки ASTM и определения фазового состава образцов;
3. индицирование линий по ожидаемым значениям параметров элементарной ячейки;
4. вычисление параметров элементарной ячейки по полученным индексам Миллера.
Уточнение параметров элементарной ячейки проводилось по стандартному методу наименьших квадратов.
Электронномикроскопические исследования полученных образцов были произведены при помощи просвечивающего электронного микроскопа JEM2000FXII (JEOL, Япония) при ускоряющем напряжении 200 кВ и увеличениях до 300000 крат. Картины электронной дифракции снимались при длине камеры 100 см.
4. Обсуждение результатов.
По данным анализов образцов, полученных керамическим синтезом, твердые растворы Bi1-xCaxOy с х = 0.1, 0.12, 0.15, 0.2 имели ромбоэдрическую структуру, подобную фазам Силлена [5] с параметрами гексагональной элементарной ячейки, приведенными в таблице 1.
Таблица 1. Значения параметров и объемов элементарной гексагональной ячейки (�) твердых растворов Bi1-xCaxOy (где а - сторона ромба, лежащего в основании элементарной ячейки, с - высота, а V - объем).
|
x=0.05 |
x=0.1 |
x=0.12 |
x=0.15 |
x=0.2 |
|
|
a |
3.958(3) |
3.939(3) |
3.948(2) |
3.946(2) |
3.932(3) |
|
с |
28.67(3) |
27.92(3) |
27.85(2) |
27.81(2) |
27.84(4) |
|
V |
388.9 |
375.2 |
375.9 |
375.0 |
372.7 |
Как видно из таблицы, с увеличением содержания кальция в твердом растворе наблюдается закономерное уменьшение объема элементарной ячейки, что вызвано меньшим ионным радиусом Ca2+, по сравнению с Bi3+.
Образец состава Bi0.75Ca0.25Oy состоял из ромбоэдрического твердого раствора и фазы Ca5Bi14O26, что согласуется с фазовой диаграммой CaO-Bi2O3. Образцы составов Bi0.98Ca0.02Oy, Bi0.95Ca0.05Oy остались неоднофазными (состоящие из моноклинного оксида висмута и ромбоэдрического твердого раствора) после трех циклов “помол-обжиг”, хотя по фазовой диаграмме данные составы относятся к области существования кубического твердого раствора. Неоднофазность образцов, по-видимому, связана с исходной неоднородностью прекурсора при керамическом синтезе. В связи с чем был проведен “бумажный синтез” данных составов.
Полученный по данной методике твердый раствор состава Bi0.95Ca0.05Oy имел ромбоэдрическую структуру с параметрами ячейки, приведенной в таблице 1. Немонотонное изменение параметров в случае этого образца, возможно, связано с его термодинамической неравновесностью. Образования кубического твердого раствора в данном случае, возможно, не наблюдалось из-за недостаточной скорости закалки образцов (закалка на охлажденной до 0С металлической плите).
Полученный твердый раствор состава Bi0.98Ca0.02Oy имел тетрагональную (искаженную кубическую) структуру с параметрами элементарной ячейки а = 7.737(6) �, с = 11.30(1) �. Для оценки скорости низкотемпературного фазового превращения была проведена серия отжигов различной длительности при температуре 550С на воздухе. Рентгенограммы данных образцов приведены на рисунке 3. Как видно из рисунка полный распад тетрагонального твердого раствора на моноклинный оксид висмута и ромбоэдрический твердый раствор происходит уже через две минуты после начала отжига при 550С. Для определения типа происходящего твердофазного превращения образец после одной минуты отжига был исследован на просвечивающем электронном микроскопе. Микрофотография и картина электронной дифракции для зоны [310] приведены на рисунках 4, 5. На микрофотографии видно образование включений произвольной формы в матрице исходного твердого раствора. При этом на электронограмме присутствуют хаотически расположенные дополнительные слабые отражения. Учитывая высокую скорость превращения при сравнительно низких температурах и морфологию образца, можно сделать предположение, что начальная стадия твердофазного распада протекает по механизму “массивного превращения” через промежуточную стадию образования термодинамически неустойчивого моноклинного твердого раствора с последующим выделением из него ромбоэдрической фазы, что подтверждается малой интенсивностью и большой шириной пиков ромбоэдрической фазы на рентгенограммах (рис.3). При больших содержаниях кальция (5 и 10 атомных %) на первой стадии распада выделяется, по-видимому, сразу ромбоэдрическая фаза.
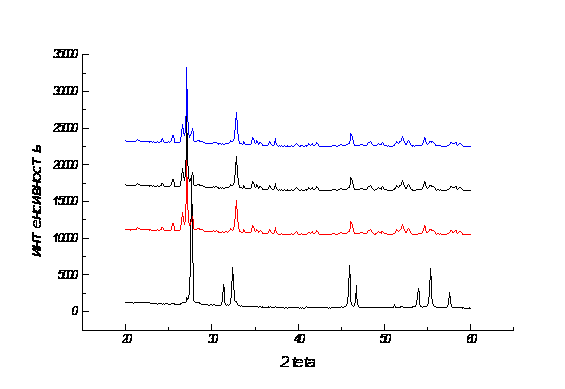
Рис.3 Рентгенограммы твердого раствора Bi0.98Ca0.02Oy после различных времен отжига при 550С (а - 1 мин, б - 2 мин, в - 5 мин, г - 15 мин). - тетрагональный твердый раствор, - моноклинный твердый раствор, - ромбоэдрический твердый раствор.
Рис. 4. Электронная микрофотография (увеличение 300000x ) твердого раствора состава Bi0.98Ca0.02Oy после одной минуты отжига при 550С.
Рис. 5. Картина электронной дифракции для зоны [310] (увеличенная в 1.76 раз) твердого раствора состава Bi0.98Ca0.02Oy после одной минуты отжига при 550С.
5. Выводы.
1. Твердые растворы Bi1-xCaxOy (х = 0.02, 0.05, 0.1, 0.12, 0.15, 0.2) были синтезированы керамическим и “бумажным” синтезами
2. Рассчитаны параметры элементарных ячеек полученных твердых растворов.
3. Были проведены ренгенографические и электронномикроскопические исследования продуктов низкотемпературного фазового распада твердого раствора Bi0.98Ca0.02Oy, на основании которых было сделано предположение о протекании начальной стадии данного процесса по механизму массивного превращения.
6. Литература.
1. Levin E.M. and Roth R.S. // J. Res. Nat. Bur. Stand, 68 A, 1964, 199.
2. Takahashi T., Iwahara H. and Nagai T. // J. Appl. Elektrochem, 2, 1972, 93.
3. Conflant P., Boivin J.C. and Tomas D. // J.Solid State Chem. 18, 1976, 133.
4. Burtor B.P., Rown C.J. and Roth R.S. // J.Res. Natl. Inst. Stand.Technol. 98, 1993, 469.
5. Sillen L.G. and Sillen B. // Z. Phys. Chem. 49B, 1944, 27.
6. Metlin Yu.G. and Tretyakov Yu.D. // J. Mater. Chem. 4(11), 1994, 1659.
7. Можаев А.П., Першин В.И., Шабатин В.П. // ЖВХО им. Д.И.Менделеeва, 34(4),1989, 504
8. Кakihana M., Yoshimura M., Masaki H., Yasuoka H., Borjersson L. // J.Appl.Phys., 71, 1992, 3904.
9. Третьяков Ю.Д., Олейников Н.Н., Можаев А.П. // “Основы криохимической технологии”, М, “Высшая школа”, 1987.
10. Жданов Г.С., Хунджуа А.Г. // “Лекции по физике твердого тела”, М, МГУ, 1988.
11. Корсаков И.Е.// дисс. канд. хим. наук, М, МГУ, 1993.