
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
Психология и педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Рефераты по сексологии
Рефераты по информатике программированию
Краткое содержание произведений
Реферат: Реакции С и О ацилирования
Реферат: Реакции С и О ацилирования
Введение.Реакции ацилирования обладают очень многими полезными свойствами. Они позволяют вести в молекулу функциональную группу C=O путем реакций присоединения либо замещения, не подвергая исходную молекулу окислению (восстановлению). Таким образом, можно получать соединения различных классов: а) амиды; б) сложные эфиры; в) ангидриды карбоновых кислот; г) кетоны и другие полезные соединения. Неудивительно, что реакции ацилирования находят широкое применение в промышленности и в химических исследованиях. В своем докладе я рассмотрю три наиболее важных типа реакций ацилирования C-ацилирование, O-ацилирование и N-ацилирование.
Реакции C-ацилирования.Наиболее часто в реакциях С-ацилирования используются металлоорганические соединения (реактивы Гриньяра, кислоты Льюиса, соединения алкила с металлом, алкоголяты металлов, а также комплексные соли с алкильными лигандами).
Реакция алкил-де-галогенирования.
Рассмотрим реакцию алкил-де-галогенирования (превращения ацилгалогенидов в кетоны с помощью металлоорганических соединений). Ацилгалогениды гладко и в мягких условиях взаимодействуют с диалкилкупратами лития, давая с высокими выходами кетоны. Это происходит по следующей схеме:
Группа R' может быть первичной, вторичной или третичной алкильной или арильной; она может содержать йодо-, кето-, нитро-, циано- и сложноэфирные группы. Успешно проведены реакции, в которых группа R была метильной, первичной алкильной и винильной. Вторичные и третичные алкильные группы можно ввести, если вместо R2CuLi использовать PhS(R)CuLi. Группа R может быть и алкинильной, если в качестве реагента применяется ацетиленид меди R’’CºCСu.
Другой тип металлоорганических реагентов, которые дают хорошие выходы кетонов при обработке ацилгалогенидами, - это кадмийорганические соединения R2Cd (получаемые из реактивов Гриньяра) В этом случае группа R может быть арильной или первичной алкильной. Вторичные и третичные алкилкадмиевые реагенты оказываются, как правило, недостаточно устойчивыми, чтобы служить полезными реагентами в этой реакции. Как в R’COX, так и в R2Cd может присутствовать сложноэфирная группа. Цинкорганические соединения ведут себя аналогично, но используются реже. Ртутьорганические соединения и тетраалкилсиланы также вступают в эту реакцию при катализе AlX3.
Реакция алкил-де-ацилокси-замещения.
Также интересна реакция алкил-де-ацилокси-замещения (превращение ангидридов, сложных эфиров и амидов карбоновых кислот в кетоны с помощью металлоорганических соединений), с помощью котрой можно получать кетоны из соединений других классов.
Как и ацилгалогениды, ангидриды и сложные эфиры карбоновых кислот при обработке реактивами Гриньяра дают третичные спирты. Для увеличения выхода кетона применяют низкие температуры и обратный порядок смешения реагентов.
Реакции сочетания ацилгалогенидов.
Известны также интересные реакции сочетания ацилгалогенидов, в результате которых можно получать симметричные a-дикетоны.
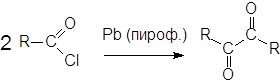 |
При действии пирофорного свинца ацилгалогениды вступают в реакцию сочетания, аналогичную реакции Вюрца:
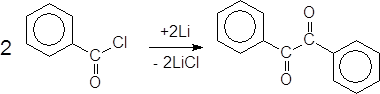 |
Аналогично проходит реакция бензоилхлорида под действием ультразвука в присутствии литиевой проволоки с образованием бензила:
Реакции ацилирования кетонов ангидридами.
Ацилирование кетонов ангидридами в присутствии трифторида бора в качестве катализатора приводит к b-дикетонам. В случае несимметричных кетонов ацилирование идет главным образом по наиболее замещенному положению:
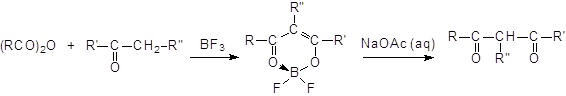
Продуктом является комплекс, содержащий BF2, который под действием водного ацетата натрия разлагается с образованием ацилированного кетона. Следовательно, трифторид бора и кетон надо брать в эквимолярных соотношениях.
Реакции ацилирования сложных эфиров сложными эфирами. Конденсации Кляйзена и Дикмана.
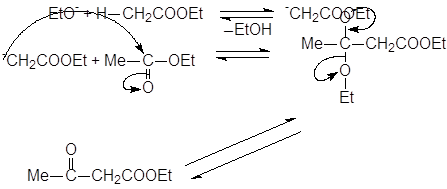 |
При обработке сложных эфиров, содержащих атом водорода в a-положении, сильным основанием (этилат натрия), происходит конденсация, приводящая к b-кетоэфирам. Эта реакция называется конденсацией Кляйзена:
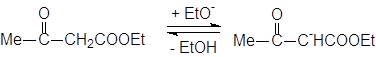 |
Необходимо отметить, что реакция идет еще дальше с образованием карбаниона и этилового спирта. Это очень важный процесс, так как он смещает равновесие вправо и позволяет получить желаемый продукт:
Когда в реакцию вводят смесь двух различных сложных эфиров, каждый из которых содержит a-атом водорода, то обычно получается смесь всех четырех возможных продуктов; вследствие этого реакция редко используется в синтетических целях. Однако, если атом водорода в a-положении имеется только в одном из сложных эфиров, смешанная реакция часто дает удовлетворительные результаты.
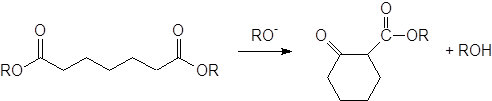 |
Если две сложноэфирные группы, участвующие в конденсации, находятся в одной молекуле, в результате получается циклический b-кетоэфир; такая реакция называется конденсацией Дикмана:
Наилучшие результаты при конденсации Дикмана получены для синтеза пяти-, шести- и семичленных циклов. Реакции, приводящие к циклам с числом атомов в кольце от 9 до 12, идут с очень низким выходом или не идут совсем; циклы большего размера синтезированы с использованием метода высокого разбавления.
Реакция ацилирования Фриделя-Крафтса.
Данная реакция позволяет присоединять к бензолу ацильный радикал. В присутствии кислот Льюиса хлорангидриды и ангидриды кислот дают ион ацилия R–C+=O, который действует как эффективный электрофильный реагент, приводя к образованию кетона:
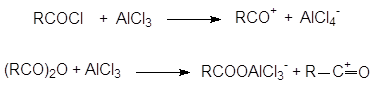
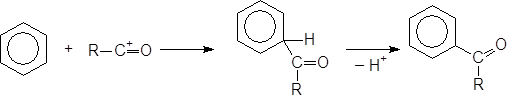
Получающийся кетон образует комплекс с хлористым алюминием извлекая его тем самым из сферы реакции. Таким образом, для завершения реакции необходимо значительно больше одного эквивалента катализатора.
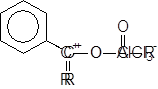 |
Имеются, однако, некоторые указания, согласно которым, если бы комплекс с AlCl3 не возникал, то кетон образовывал бы комплекс с ионом ацилия, который не мог бы атаковать основной субстрат, в данном случае бензол.
Причем если радикал имеет сильно разветвленное строение, то может происходить отщепление C=O, и в этом случае вместо ожидаемого ацилирования будет наблюдаться алкилирование субстрата:
Интересным примером использования реакции Фриделя-Крафтса может быть следующая двухстадийная и весьма важная в синтетическом отношении реакция:
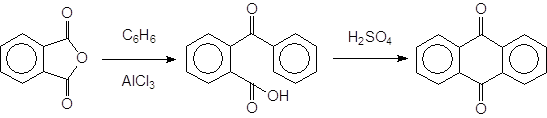 |
Реакции N-ацилирования.
Ацилирование аминов ацилгалогенидами.
Реакции амино-де-галогенирования наиболее часто используются для синтеза амидов. Действие аммиака или аминов на ацилгалогениды представляет собой общий метод синтеза амидов.
Реакция сильно экзотермична и требует тщательного контроля, обычно охлаждением или разбавлением. При использовании аммиака получают незамещенные амиды, из первичных аминов получают N-замещенные амиды, а из вторичных аминов – N,N-дизамещенные амиды. Аналогично можно ацилировать ариламины. В некоторых случаях для связывания выделяющейся галогеноводородной кислоты добавляют водный раствор щелочи. Такая реакция носит название метода Шоттена-Баумана.
Гидразин и гидроксиламин также реагируют с ацилгалогенидами, давая соответственно гидразиды RCONHNH2 и гидроксамовые кислоты RCONHOH; эта реакция часто используется для синтеза данных соединений. Если вместо ацилгалогенида взять фосген, то как ароматические, так и алифатические первичные амины дают хлороформамиды ClCONHR, которые теряя HCl, превращаются в изоцианаты RNCO. Это один из наиболее распространенных методов синтеза изоцианатов.
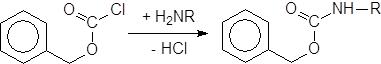 |
Тиофосген при аналогичной обработке дает изотиоцианаты. Фосген в этой реакции можно заменить более безопасным трихлорометилхлороформиатом. При действии первичных аминов на хлороформиаты ROCOCl получаются карбаматы ROCONHR’. Примером этой реакции служит защита аминогруппы в аминокислотах и пептидах действием карбобензоксихлорида:
Аминогруппы вообще часто защищают превращением в более устойчивые – амидные. Взаимодействие ацилгалогенидов с нитридом лития дает N,N-диациламиды (триациламины).
Ацилирование аминов ангидридами.
По механизму и диапазону применимости реакция амино-де-ацилокси-замещения аналогична реакции описанной в предыдущем разделе и может быть проведена с участием аммиака, первичных или вторичных аминов.
Однако при использовании аммиака и первичных аминов получаются также и имиды, в которых с атомом азота свяэаны две ацильные группы. Это происходит особенно легко в случае циклических ангидридов, из которых образуются циклические имиды:
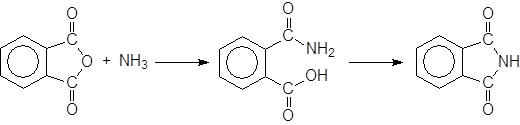
Второй стадией этой реакции, которая намного медленне первой, является атака атома азота амидной группы на карбоновую кислоту.
Ацилирование аминов карбоновыми кислотами.
При обработке карбоновых кислот аммиаком или аминами получаются соли. Соли, полученные из аммиака, а также первичных и вторичных аминов в результате пиролиза дают амиды, но этот метод менее удобен, чем реакции аминов с ангидридами, ацилгалогенидами и сложными эфирами, и редко используется в препаративных целях.
Хотя и взаимодействие кислот с аминами не приводит непосредственно к амидам, можно добиться чтобы эта реакция шла с хорошим выходом при комнатной или немного повышенной температуре.
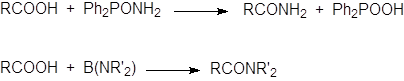 |
Кислоты можно превратить в амиды также нагреванием с амидами других карбоновых кислот (обмен), сульфоновых или фосфиновых кислот или действием трис(алкиламино)боранов [B(NHR’)3] или трис(диалкиламино)боранов [B(NR’2)3]:
Ацилирование аминов сложными эфирами.
Превращение сложных эфиров в амиды – полезный метод синтеза незамещенных, N-замещенных и N,N-дизамещенных амидов из соответствующих аминов.
Реакцию можно проводить с алкильными или ароматическими группами R и R’. Особенно хорошей уходящей группой является n-нитрофенильная. Эта реакция весьма ценна, так как многие сложные эфиры легкодоступны или сравнительно легко получаются даже в тех случаях, когда этого нельзя сказать о соответствующем ангидриде кислоты или ацилгалогениде. Как и по реакции с ацилгалогенидами, этим методом из сложных эфиров можно синтезировать гидразиды и гидроксамовые кислоты действием гидразина и гидроксиламина соответственно. И гидразин, и гидроксиламин взаимодействуют быстрее, чем аммиак или первичные амины. Вместо сложных эфиров часто используют фенилгидразиды, получаемые из фенилгидразина.
Остаётся добавить, что реакция образования гидроксамовых кислот. которые в присутсвии трёхвалентного железа дают окрашенные комплексы, часто используется как тест на сложные эфиры.
Ацилирование аминов амидами.
Это реакция обмена, и ее обычно проводят с солью амина. Уходящей группой служит, как правило, NH2, а не NHR или NR2; в качестве реагентов наиболее широко применяются первичные амины (в виде солей).
Для образования комплекса с уходящим аммиаком можно добавлять BF3. Эту реакцию часто применяют для получения замещенных производных мочевины из самой мочевины:
Диметилформамид можно превратить в другие формамиды продолжительным нагреванием с первичным или вторичным амином.
Реакции O-ацилирования.Гидролиз ацилгалогенидов.
Ацилгалогениды очень реакционноспособны, поэтому гидролиз проходит легко. Большинство галогеноангидридов простых кислот следует хранить в безводных условиях, так как они реагируют с влагой воздуха. Поэтому обычно вода оказывается достаточно сильным нуклеофилом для проведения реакции гидролиза, хотя в отдельных случаях необходимо использовать гидроксид-ион.
Реакция, как правило, не имеет синтетической ценности, так как ацилгалогениды обычно получают из кислот. Реакционная способность ацилгалогенидов изменяется в следующем ряду: F<Cl<Br<I. При использовании в качестве нуклеофила карбоновой кислоты возможна реакция обмена.
Гидролиз галогеноангидридов обычно не катализируется кислотами, за исключением ацилфторидов, когда образование водородной связи может способствовать отщеплению фтора.
Гидролиз ангидридов.
Гидролиз ангидридов протекает несколько труднее, чем гидролиз галогеноангидридов, но и в этом случае вода обычно оказывается достаточно сильным нуклеофилом.
Гидролиз ангидридов можно катализировать основаниями. Конечно, OH-группа атакует более энергично, чем вода, но и другие основания могут катализировать эту реакцию. Это явление, называется нуклеофильным катализом.
Гидролиз сложных эфиров.
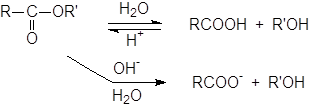 |
Гидролиз сложных эфиров обычно катализируется как кислотами, так и основаниями. Поскольку группа OR обладает более слабыми нуклеофугными свойствами, чем галогены или OCOR, вода не гидролизует большинство сложных эфиров.
При катализе основаниями, атакующей частицей служит более сильный нуклеофил – OH-группа. Эта реакция носит название омыления и приводит к соли кислоты. Кислоты катализируют реакцию за счет того, что положительный заряд атома углерода карбонильной группы становится больше, и, следовательно, он легче подвергается атаке нуклеофилом. Обе реакции обратимы, и поэтому практической ценностью обладают только тогда, когда равновесия удаётся каким либо образом сместить вправо. А поскольку образование соли – один из таких способов, гидролиз сложных эфиров в препаративных целях почти всегда проводят в щелочных растворах, за исключением тех случаев, когда вещество неустойчиво к действию оснований.
Гидролиз амидов.
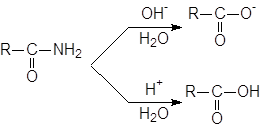 |
Незамещенные амиды RCONH2 способны гидролизоваться под действием как кислотных, так и основных катализаторов; при этом образуются соответствующая свободная кислота и ион аммония или соль кислоты и аммиак.
Аналогично можно гидролизовать N-замещенные (RCONHR’) и N,N-дизамещенные (RCONR’2) амиды, причем вместо аммиака получаются первичные и вторичные амины соответственно (или их соли). Вода является слишком слабым нуклеофилом для гидролиза большинства амидов, таккак группа NH2 обладает еще более низкими нуклеофугными свойствами, чем группа OR. Даже в условиях кислотного или основного катализа часто требуется длительное нагревание.
Алкоголиз ацилгалогенидов.
Реакция ацилгалогенидов со спиртами – наилучший общий метод получения сложных эфиров. Реакция находит широкое применение и может быть проведена для субстратов, содержащих различные функциональные группы.
Для связывания образующегося НХ часто добавляют основания. В методе Шоттена-Баумана используется водный раствор щелочи, но часто применяется пиридин. Как R, так и R’ могут быть первичными, вторичными или третичными алкилами или арилами. В сложных случаях, особенно для стерически затрудненных кислот или третичных R’, вместо спирта можно брать алкоголят ион.
При использовании в качестве ацилгалогенидов фосгена можно получить галоформные эфиры или карбонаты:
Важный пример – это синтез из фосгена и бензилового спирта карбобензоксихлорида (PhCH2OCOCl), который широко применяется для защиты аминогрупп в пептидном синтезе.
Алкоголиз ангидридов.
Диапазон применяемости этого метода такой же, как и реакции алкоголиза ацилгалогенидов. И хотя ангидриды немного менее реакционно способны, чем ацилгалогениды, их часто используют для получения сложных эфиров.
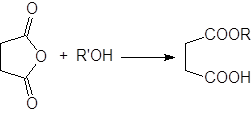 |
В качестве катализаторов применяют кислоты, кислоты Льюиса и основания, но наиболее часто – пиридин. Реакция циклических ангидридов приводит к моноэтерифицированным дикарбоновым кислотам, например:
Этерификация кислот.
Этерификация кислот спиртами представляет собой реакцию, обратную реакции гидролиза сложных эфиров:
Ее можно осуществить только тогда, когда равновесие удается сместить вправо. Для этой цели имеется много способов, среди которых:
1) прибавление одного из реагентов (обычно спирта) в избытке;
2) удаление эфира или воды отгонкой;
3) азеотропная отгонка воды;
4) удаление воды, используя водоотнимающие средства или молекулярные сита.
Если R’ = метил, то наиболее общий способ смещения равновесия – это добавление избытка МеОН, а если R’ = этил, то предпочтительнее удалять воду азеотропной отгонкой. В качестве катализаторов чаще всего используются серная кислота и TsOН, хотя в случае некоторых активных кислот (например муравьиной или трифтороуксусной) катализатора не требуется. Группа R’ может быть не только метильной или этильной, но также и другой первичной или вторичной алкильной группой, однако третичные спирты обычно образуют карбокатионы и происходит элиминирование. для получения эфиров фенолов можно использовать сами фенолы, но выходы, как правило очень низкие. Этерификация катализируется кислотами (но не основаниями).
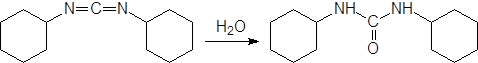 |
Другим способом получения сложных эфиров из кислоты является обработка спиртом в присутствии дегидратирующих веществ, одним из которых является дициклогексилкарбодиимид, в ходе реакции превращающийся в дициклогексилмочевину:
Алкоголиз сложных эфиров. Переэтерификация.
Переэтерификация катализируется кислотами или основаниями. Это обратимая реакция и равновесие необходимо смещать в желаемую сторону.
Во многих случаях низкокипящие эфиры можно превратить в более высококипящие путем отгонки низкокипящего спирта по мере его образования. Эта реакция была использована как метод ацилирования первичных ОН-групп в присутствии вторичных ОН- групп.
Ацилирование карбоновых кислот ацилгалогенидами.
Несимметричные, а также и симметричные ангидриды часто получают обработкой ацилгалогенидов с солью кислоты.
Катионами в этой соли служат Na+,K+ и Ag+, но чаще пиридин или другой третичный амин прибавляют к кислоте и полученную таким образом соль обрабатывают ацилгалогенидом. Соли таллия особенно эффективны и взаимодействуют с ацилгалогенидами, давая ангидриды с высокими выходами. Симметричные ангидриды синтезируют по реакции ацилгалогенида с водным раствором NaOH в условиях межфазного катализа.
Ацилирование карбоновых кислот кислотами.
Из двух молекул обычной кислоты ангидрид образуется только в присутствии дегидратирующего агента, смещающего равновесие в этой реакции вправо.
Наиболее распространенными дегидратирующими агентами являются уксусный и трифтороуксусный ангидриды, дициклогексилкарбодиимид, метоксиацетилен и P2O5. Этот метод дает плохие результаты при получении смешанных ангидридов, которые во всех случаях обычно диспропорционируют на два простых ангидрида при нагревании. Однако простое нагревание дикарбоновых кислот приводит к циклическим ангидридам при условии, что в образующемся цикле содержатся пять, шесть или семь атомов. Малоновая кислота и ее производные, которые должны давать четырехчленные циклические ангидриды, не вступают в эту реакцию при нагревании, а декарбоксилируются: Список литературы.
1)”Органическая химия” Джерри Марч; Москва “Мир” 1987;
2)”Механизмы реакций в органической химии” Питер Сайкс;
“Химия” 1971;
3)”Органическая химия” Ю.С.Шабаров; “Химия” 1994;
4)”Основы органической химии” Франк Л. Вайзман; “Химия” 1995.
Оглавление
Введение. 1
Реакции C-ацилирования. 1
Реакции сочетания ацилгалогенидов. 2
Реакции ацилирования кетонов ангидридами. 2
Реакции ацилирования сложных эфиров сложными эфирами. Конденсации Кляйзена и Дикмана. 3
Реакция ацилирования Фриделя-Крафтса. 4
Реакции N-ацилирования. 5
Ацилирование аминов ацилгалогенидами. 5
Ацилирование аминов ангидридами. 6
Ацилирование аминов карбоновыми кислотами. 6
Ацилирование аминов сложными эфирами. 6
Ацилирование аминов амидами. 7
Реакции O-ацилирования. 7
Гидролиз ацилгалогенидов. 7
Гидролиз ангидридов. 8
Гидролиз сложных эфиров. 8
Гидролиз амидов. 8
Алкоголиз ацилгалогенидов. 9
Алкоголиз ангидридов. 9
Этерификация кислот. 10
Алкоголиз сложных эфиров. Переэтерификация. 10
Ацилирование карбоновых кислот ацилгалогенидами. 10
Ацилирование карбоновых кислот кислотами. 11
Список литературы. 11